Way to predict Drug-target interaction binding affinity using Convolution Neural Network in Drug Discovery Pipeline
In this blog, I am writing about use of deep learning architecture for identification of drug-target interactions (DTI) strength (binding affinity) using character based sequence model.
As the field of drug discovery expands with the discovery of new drugs, re-purposing of existing drugs and identification of novel interacting partners for approved drugs is also gaining interest, since identification of novel drug-target (DT) interactions is a substantial part of the drug discovery process there are many existing model to predict the drug-target interaction based on binary classification but in that constrain is the data sets constitutes a major problem, since negative (non-binding ) information is hard to find there. This article is about an approach to predict the binding affinities of protein-ligand interactions with deep learning models using only sequences (1D representations) of proteins and ligand. It is a CNN based deep learning model that employ chemical and biological textual sequence information to predict binding affinity (Binding affinity provide information on the strength of the interaction between a drug-target (DT) pair).
Data-sets:
Model evaluation is done on KIBA data-set (kinase inhibitors bio-active data) in which kinase inhibitor bio-activities from different sources were combined. The KIBA data set originally comprised 229 proteins and 2111 drugs but due to our resource constrains(i.e. for more data to perform it requires GPU environment but we are using CPU environment) we filtered the data-set based on length of ligands and proteins. The constrains of length of every ligand sequence is 50(characters) and for every protein sequence it is 600(characters), now our data-set contains 111 proteins and 810 ligands and corresponding affinity values.
Data Input representation to the model:
We used one-hot encoding that uses binary integers (0, 1) for the ligand (characters) and protein (characters) to represent inputs Since Both SMILES (Len<50) and protein sequences (Len<600) have varying lengths. Hence, in order to create an effective representation form (one-hot encoding), we decided to fixed maximum lengths of 50(characters) for SMILES and 600(characters) for protein sequences in our data-set. Final one-hot representation of ligands and proteins having dimension of (62x50) and (25x600) respectively. Here 62 and 25 are unique characters from SMILES (ligand) and proteins respectively. For more details refer this article:: href=”https://arxiv.org/abs/1801.10193”
Model Preparation:
In this article we treated protein-ligand interaction prediction as a regression problem by aiming to predict the binding affinity scores and used deep learning architecture, Convolutional Neural Network (CNN). For this problem set (i.e. for ligands and proteins) we build a CNN-based prediction model that comprises two separate CNN blocks, each of which aims to learn representations from SMILES strings and protein sequences separately. For each CNN block the number of filters is 16 in first and 32 in the second convolutional layer then followed by the max-pooling layer. The final features of the max-pooling layers were concatenated and fed into two FC layers with a dropout layer (rate=0.15), which we named as DeepDTA.
- Activation function used: Rectified Linear Unit (ReLU).
- Loss function used: Root mean squared error (RMSE).
The proposed model that combines two CNN blocks is illustrated below:
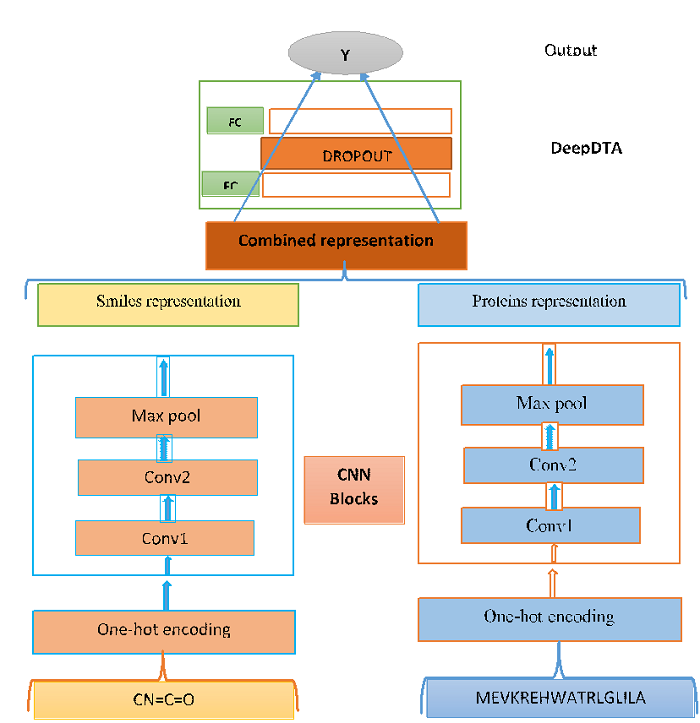
CNN based DeepDTA model
#########MODEL
import data
from data import*
import torch
import torch.nn as nn
from torch import optim
import torch.nn.functional as F
class CNNcom(nn.Module):
def __init__(self):
super(CNNcom, self).__init__()
# for smiles
self.sconv1 = nn.Conv1d(in_channels=62, out_channels=16, kernel_size=2, stride=1, padding=1)
self.sconv2 = nn.Conv1d(16, 32, 2, stride=1, padding=1)
self.pool = nn.MaxPool1d(2, 2)
#for proteins
self.pconv1 = nn.Conv1d(in_channels=25, out_channels=16, kernel_size=2, stride=1, padding=1)
self.pconv2 = nn.Conv1d(16, 32, 2, stride=1, padding=1)
self.dropout = nn.Dropout(0.15)
self.linear1 = nn.Linear(5216, 256) # put the z
self.linear2 = nn.Linear(256, 1)
def forward(self, x, x1):
x = self.pool(F.relu(self.sconv1(x)))
x = self.pool(F.relu(self.sconv2(x)))
x1 = self.pool(F.relu(self.pconv1(x1)))
x1 = self.pool(F.relu(self.pconv2(x1)))
x = x.view(-1, 13*32)
x1 = x1.view(-1,150*32)
x2 = torch.cat([x, x1],1)
x2 = F.relu(self.linear1(x2))
x2 = self.dropout(x2)
x2 = self.linear2(x2)
return x2
model2 = CNNcom()
if __name__ == '__main__':
print(model2)
Training:
For training and loss calculation we used Adam optimizer and RMSE loss function respectively. for more details visit this github link:-
Conclusion:
when two CNN-blocks that learn representation of proteins and drugs based on raw sequence data are used in conjunction with DeepDTA, the performance is significantly considerable.